GORTEC 2024-01 - TATIANA
Etude de phase II évaluant TPEx (taxotere-cisplatin-cetuximab) suivi d’un traitement demaintenance par Avelumab et Cetuximab en première ligne de traitement des patients ayant uncarcinome épidermoïde de la tête et du cou métastatique ou en récidive
Phase : II
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Détails de l'essai
Objectif principal
Survie globale.
Objectif(s) secondaire(s)
Estimer l’efficacité en terme de survie sans progression (SSP).
Evaluer la sécurité du traitement (chimiothérapie d’induction et association avelumab – cetuximab comme maintenance après le schéma TPEx).
Résumé / schéma de l'étude
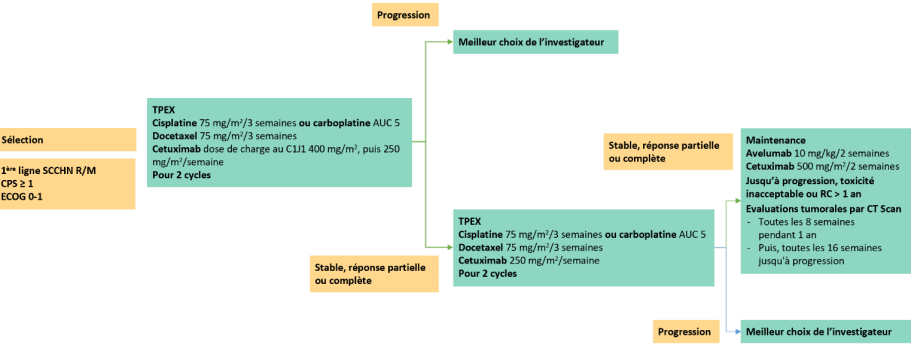
Critère(s) d'inclusion
- Hommes et femmes adultes ≥ 18 ans et < 75 ans.
- Carcinome épidermoïde de la tête et du cou (SCCHN) récurrent et/ou métastatique confirmé histologiquement (cavité buccale, pharynx, larynx), ne se prêtant pas à un traitement local à visée curative (chirurgie ou radiothérapie avec ou sans chimiothérapie) ; carcinome épidermoïde sans primitif connu si HPV positif.
- Détection de l’expression de la protéine PD-L1 dans des échantillons de tissus SCCHN fixés au formol et inclus en paraffine (FFPE) déterminés par un score positif combiné (CPS) ≥ 1 à l’aide d’un test IHC local.
- Score de performance de l’Eastern Cooperative Oncology Group (ECOG) ≤ 1.
- Patients sans contre-indication au TPEx (avec cisplatine ou carboplatine), au docétaxel, au cétuximab et à l’immunothérapie (avelumab). Les investigateurs doivent se référer aux dernières versions des Résumés des Caractéristiques des Produits (RCP).
- Documentation du statut de p16 en tant que substitut du statut du virus du papillome humain (HPV) pour le carcinome épidermoïde de l’oropharynx.
- Maladie mesurable par tomodensitométrie ou IRM selon les critères RECIST 1.1.
- En cas de radiothérapie sans traitement systémique, la radiothérapie curative préalable doit être terminée au moins 4 semaines avant l’administration de TPEx et/ou la radiothérapie palliative préalable doit être terminée au moins 2 semaines avant l’administration de TPEx.
- Les valeurs de laboratoire de screening doivent répondre aux critères suivants (en utilisant NCI-CTCAE v5) et doivent être obtenues dans les 14 jours précédant la vérification de l’éligibilité :
- Globules blancs > 2 000/μL
- Neutrophiles polynucléaires >1,5 x 109/L
- Plaquettes > 100 x 109/L
- Hémoglobine > 9,0 g/dL
- ALAT/ASAT < 3,0 x LSN en l’absence de métastases hépatiques ou < 5 x LSN en présence de métastases hépatiques
- Bilirubine < 1,5 x LSN (sauf syndrome de Gilbert : < 3,0 mg/dL)
- Clairance de la créatinine > 60 mL/min (mesurée ou calculée de préférence par la formule de Cockcroft et Gault) pour l’administration de cisplatine et clairance de la créatinine entre ≥ 40 mL/min et ≤ 60 mL/min (mesurée ou calculée de préférence par la formule de Cockcroft et Gault) pour l’administration de carboplatine .
- Niveaux de calcium normalisés et maintenus dans les limites normales pour l’entrée dans l’étude. La gestion médicale des niveaux de calcium est autorisée. Remarque : les niveaux normaux de calcium peuvent être basés sur le calcium ionisé ou ajustés en fonction de l’albumine.
- Rétablissement au niveau de baseline ou ≤ Grade 1 selon les critères de terminologie communs pour les événements indésirables (CTCAE) v5 d’un ou plusieurs EI liés à des traitements antérieurs, à moins que les EI ne soient jugés cliniquement non significatifs par l’investigateur et/ou stables avec un traitement de soutien.
- Femmes en âge de procréer doivent réaliser un test de grossesse sérique ou urinaire négatif lors du contrôle d’éligibilité. Le test de grossesse doit être effectué dans les 72 heures
précédant la vérification de l’éligibilité. - Hommes et les femmes (en âge de procréer) sexuellement actifs doivent accepter d’utiliser des méthodes contraceptives hautement efficaces depuis la signature de l’ICF jusqu’à au
moins 7 mois après le traitement pour les femmes et 4 mois après le traitement pour les hommes. - Femmes allaitante doivent arrêter d’allaiter avant la vérification de l’éligibilité.
- Consentement éclairé écrit, signé et daté avant toute procédure, échantillonnage et analyse spécifiques à l’étude.
- Patients disposant d’une couverture d’assurance sociale.
- Patients doivent être disposés et capables de se conformer aux visites programmées, aux tests de laboratoire du plan de traitement et aux autres procédures d’étude.
Critère(s) de non-inclusion
- Chimiothérapie systémique antérieure pour le carcinome de la tête et du cou, sauf si elle est administrée dans le cadre d’un traitement multimodal pour une maladie localement avancée qui a été achevée plus de 6 mois avant l’entrée à l’étude.
- Carcinomes récidivants ou métastatiques du nasopharynx confirmés histologiquement. Les carcinomes épidermoïdes de primitif inconnu HPV négatif ou les glandes salivaires ou les
histologies non squameuses (par exemple, mélanome muqueux) ne sont pas autorisés. - Tout trouble médical grave ou incontrôlé qui, de l’avis de l’investigateur, peut augmenter le risque associé à la participation à l’étude ou à l’administration du médicament à l’étude, altérer la capacité du sujet à recevoir un protocole thérapeutique ou interférer avec l’interprétation des résultats de l’étude.
- Antécédents de tumeur maligne active au cours des 3 années précédentes, à l’exception des cancers localement curables qui ont été apparemment guéris, comme le cancer de la peau
basocellulaire ou épidermoïde, le cancer superficiel de la vessie ou le carcinome in situ de la prostate, du col de l’utérus ou du sein. - Patients atteints d’une maladie auto-immune active, connue ou suspectée. Patients atteints de diabète de type I stabilisé sous traitement, d’hypothyroïdie résiduelle due à une maladie auto-immune nécessitant uniquement un remplacement hormonal, un psoriasis ne nécessitant pas de traitement systémique ou des conditions qui ne devraient pas se
reproduire en l’absence d’un déclencheur externe sont autorisés. - Patients présentant une affection nécessitant une administration systémique chronique de corticostéroïdes (> 10 mg d’équivalents prednisone par jour) ou d’autres médicaments
immunosuppresseurs dans les 14 jours suivant la vérification de l’éligibilité. Les stéroïdes inhalés ou topiques et les doses de remplacement surrénalien > 10 mg par jour d’équivalents prednisone sont autorisés en l’absence de maladie auto-immune active. - Patients ayant déjà reçu un traitement par anti-PD1, anti-PD-L1 (ou tout autre anticorps ou médicament ciblant spécifiquement la costimulation des lymphocytes T ou les voies des
checkpoint). - Traitement anti-EGFR antérieur reçu moins de 6 mois avant l’inclusion à l’essai.
- Patients avec un test positif connu pour le virus de l’immunodéficience humaine (VIH) ou syndrome d’immunodéficience acquise (SIDA).
- Patients avec des tests positifs connus pour l’antigène de surface du virus de l’hépatite B (AgHBV) ou acide ribonucléique du virus de l’hépatite C (ARN du VHC) indiquant une infection active ou chronique.
- Utilisation de vaccins non oncologiques contenant des virus ou des bactéries vivants pour la prévention des maladies infectieuses dans les 4 semaines précédant la vérification de
l’éligibilité. L’utilisation du vaccin inactivé contre la grippe saisonnière est autorisée. - Antécédents de réaction d’hypersensibilité sévère à tout anticorps monoclonal humain.
- Conditions psychiatriques concomitantes graves qui limiteraient le respect des exigences de l’étude.
- Infection systémique grave nécessitant un traitement hospitalier avec des antibiotiques intraveineux dans les 14 jours précédant la vérification de l’éligibilité.
- Antécédents de transplantation d’organes.
- Patients présentant une tumeur hémorragique, uniquement si administration du Carboplatine.
- Traitement concomitant par phénytoïne and fosphénytoïnes durant le traitement à l’étude.
- Antécédents d’allergie à la viande rouge ou aux morsures de tiques ou résultats positifs des tests d’anticorps IgE contre le cetuximab (α-1-3-galactose).
- Maladie pulmonaire interstitielle.
- Antécédents de maladie cardiaque incontrôlée ou symptomatique.
- Personne privée de liberté par décision judiciaire ou administrative, ou sous toute forme de tutelle ou de curatelle.
Calendrier prévisionnel
Lancement de l’étude : Janvier 2025
Fin estimée des inclusions : Novembre 2025
Nombre de patients à inclure : 70
Coordonnateur de l'étude
Dr Caroline EVEN – Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
Groupe d’Oncologie Radiothérapie Tête et Cou (GORTEC)
Dernière mise à jour le 9 avril 2025